Convergence of the optical spectra¶
In the previous Optical Spectra from TD we have seen how to obtain the absorption spectrum of a molecule. Here we will perform a convergence study of the spectrum with respect to the grid parameters using again the methane molecule as an example.
import matplotlib.pyplot as plt
from pathlib import Path
from postopus import Run
import subprocess
from multiprocessing import Pool
Convergence with the spacing¶
In the Total energy convergence tutorial we found out that the total energy of the methane molecule was converged to within 0.1 eV for a spacing of 0.18 Å and a radius of 3.5 Å. We will therefore use these values as a starting point for our convergence study. Like for the total energy, the only way to check if the absorption spectrum is converged with respect to the spacing is by repeating a series of calculations, with identical input files except for the grid spacing. The main difference in this case is that each calculation for a given spacing requires several steps:
Ground-state calculation,
Three time-dependent propagations with perturbations along “x”, “y” and “z”,
Calculation of the spectrum using the
oct-propagation_spectrumutility.
As we have seen before, the \(T_d\) symmetry of methane means that the spectrum is identical for all directions, so in this case we only need to perform the convergence study with a perturbation along the “x” direction.
All these calculations can be done by hand, but we would rather use a script for this boring an repetitive task. We will then need the following file.
inp¶
This file is the same as the one used in the previous Optical Spectra from TD:
inp_template_spacing = """
stdout = 'stdout_{calculation_mode}_{spacing_value}.txt'
stderr = 'stderr_{calculation_mode}_{spacing_value}.txt'
CalculationMode = {calculation_mode}
UnitsOutput = eV_angstrom
Eigensolver = chebyshev_filter
ExtraStates = 4
Radius = 3.5*angstrom
Spacing = {spacing_value}*angstrom
CH = 1.097*angstrom
%Coordinates
"C" | 0 | 0 | 0
"H" | CH/sqrt(3) | CH/sqrt(3) | CH/sqrt(3)
"H" | -CH/sqrt(3) |-CH/sqrt(3) | CH/sqrt(3)
"H" | CH/sqrt(3) |-CH/sqrt(3) | -CH/sqrt(3)
"H" | -CH/sqrt(3) | CH/sqrt(3) | -CH/sqrt(3)
%
TDPropagator = aetrs
TDTimeStep = 0.0023/eV
TDMaxSteps = 4350 # ~ 10.0/TDTimeStep
TDDeltaStrength = 0.01/angstrom
TDPolarizationDirection = 1
"""
This loop performs the three steps mentioned above for a given list of spacings. You will notice that we are not including smaller spacings than 0.18 Å. This is because we know from previous experience that the optical spectrum is well converged for spacings larger than the one required to converge the total energy.
Running this calculation will take some time, so you might want to have a break.
spacing_list = [0.26, 0.24, 0.22, 0.20, 0.18]
def run_octopus_for_spacing(spacing):
runpath = Path(f"2_Convergence_of_optical_spectra/variable_spacing/run_{spacing}")
runpath.mkdir(exist_ok=True, parents=True)
# Run gs calculation
(runpath / "inp").write_text(inp_template_spacing.format(calculation_mode="gs", spacing_value=spacing))
subprocess.run(f"cd {runpath} && octopus", shell=True)
# Run td calculation
(runpath / "inp").write_text(inp_template_spacing.format(calculation_mode="td", spacing_value=spacing))
subprocess.run(f"cd {runpath} && octopus", shell=True)
# Run the oct-propagation_spectrum utility
(runpath / "inp").write_text("""
UnitsOutput = eV_angstrom
stdout='stdout_propagation_spectrum.txt'
stderr='stderr_propagation_spectrum.txt'
"""
)
subprocess.run(f"cd {runpath} && oct-propagation_spectrum", shell=True)
# Run octopus for all spacings parallel (see also https://docs.python.org/3/library/multiprocessing.html)
with Pool(5) as pool:
pool.map(run_octopus_for_spacing, spacing_list)
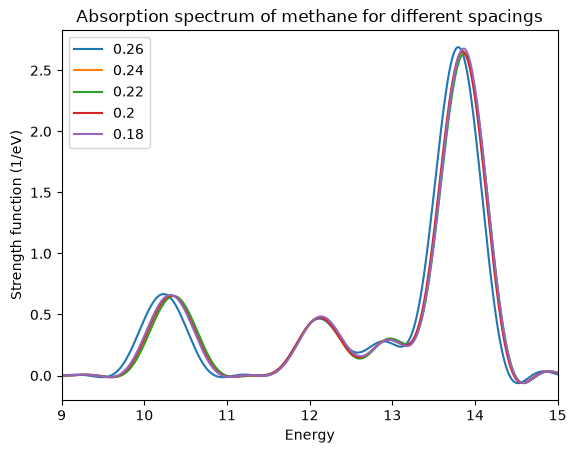
Once the loop is finished running, we need to compare the spectra for the different spacings. You can see below how this plot should look like. To make the plot easier to read, we have restricted the energy range. From the plot it is clear that a spacing of 0.24 Å is sufficient to have the position of the peaks converged to within 0.1 eV.
fig, ax = plt.subplots()
ax.set_title("Absorption spectrum of methane for different spacings")
ax.set_xlabel("Energy (eV)")
ax.set_ylabel("Strength function (1/eV)")
for spacing in spacing_list:
run = Run(f"2_Convergence_of_optical_spectra/variable_spacing/run_{spacing}")
cross_section_vector = run.td.cross_section_vector()
cross_section_vector.plot(
x="Energy", y="StrengthFunction(1)", ax=ax, label=f"{spacing}", xlim=[9, 15]
)
Convergence with the radius¶
We will now study how the spectrum changes with the radius. We will proceed as for the spacing, by performing a series of calculations with identical input files except for the radius. For this we will use the following file.
inp¶
This file is the same as the prevous one, with the exception of the spacing and the time-step. As we have just seen, a spacing of 0.24 Å is sufficient, which in turns allows us to use a larger time-step.
inp_template_radius = """
stdout = 'stdout_{calculation_mode}_{radius_value}.txt'
stderr = 'stderr_{calculation_mode}_{radius_value}.txt'
CalculationMode = {calculation_mode}
UnitsOutput = eV_angstrom
Radius = {radius_value}*angstrom
Spacing = 0.24*angstrom
CH = 1.097*angstrom
%Coordinates
"C" | 0 | 0 | 0
"H" | CH/sqrt(3) | CH/sqrt(3) | CH/sqrt(3)
"H" | -CH/sqrt(3) |-CH/sqrt(3) | CH/sqrt(3)
"H" | CH/sqrt(3) |-CH/sqrt(3) | -CH/sqrt(3)
"H" | -CH/sqrt(3) | CH/sqrt(3) | -CH/sqrt(3)
%
TDPropagator = aetrs
TDTimeStep = 0.004/eV
TDMaxSteps = 2500 # ~ 10.0/TDTimeStep
TDDeltaStrength = 0.01/angstrom
TDPolarizationDirection = 1
"""
The loop works as the loop above, changing the radius instead of the spacing. This calculation again will take some minutes.
list_of_radii = [3.5, 4.5, 5.5, 6.5, 7.5]
def run_octopus_for_radius(radius):
runpath = Path(f"2_Convergence_of_optical_spectra/variable_radius/run_{radius}")
runpath.mkdir(exist_ok=True, parents=True)
# Run gs calculation
(runpath / "inp").write_text(inp_template_radius.format(calculation_mode="gs", radius_value=radius))
subprocess.run(f"cd {runpath} && octopus", shell=True)
# Run td calculation
(runpath / "inp").write_text(inp_template_radius.format(calculation_mode="td", radius_value=radius))
subprocess.run(f"cd {runpath} && octopus", shell=True)
# Run the oct-propagation_spectrum utility
(runpath / "inp").write_text("""
UnitsOutput = eV_angstrom
stdout='stdout_propagation_spectrum.txt'
stderr='stderr_propagation_spectrum.txt'
"""
)
subprocess.run(f"cd {runpath} && oct-propagation_spectrum", shell=True)
# Run octopus for all spacings parallel (see also https://docs.python.org/3/library/multiprocessing.html)
with Pool(5) as pool:
pool.map(run_octopus_for_radius, list_of_radii)
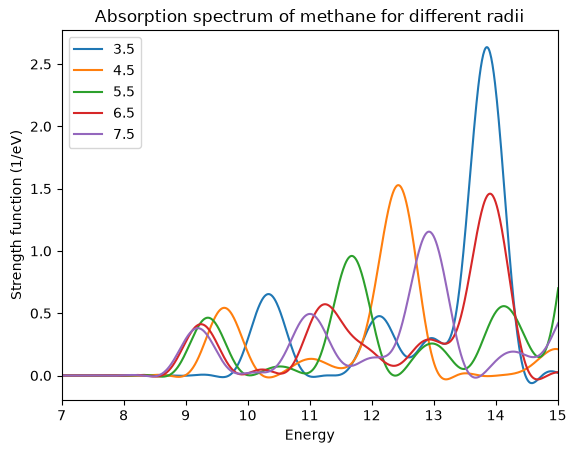
We now plot the spectra from the cross_section_vector files. The plot shows that in this case the changes are quite dramatic. To get the first peak close to 10 eV converged within 0.1 eV a radius of 6.5 Å is necessary. The converged transition energy of 9.2 eV agrees quite well with the experimental value of 9.6 eV and with the TDDFT value of 9.25 eV obtained by other authors.\(^1\)
fig, ax = plt.subplots()
ax.set_title("Absorption spectrum of methane for different radii")
ax.set_xlabel("Energy (eV)")
ax.set_ylabel("Strength function (1/eV)")
for radius in list_of_radii:
run = Run(f"2_Convergence_of_optical_spectra/variable_radius/run_{radius}")
cross_section_vector = run.td.cross_section_vector()
cross_section_vector.plot(
x="Energy", y="StrengthFunction(1)", ax=ax, label=f"{radius}", xlim=[7, 15]
);
What about the peaks at higher energies? Since we are running in a box with zero boundary conditions, we will also have the box states in the spectrum. The energy of those states will change with the inverse of the size of the box and the corresponding peaks will keep shifting to lower energies. However, as one is usually only interested in bound-bound transitions in the optical or near-ultraviolet regimes, we will stop our convergence study here. As an exercise you might want to converge the next experimentally observed transition. Just keep in mind that calculations with larger boxes might take quite some time to run.
References¶
N. N. Matsuzawa, A. Ishitani, D. A. Dixon, and T. Uda, Time-Dependent Density Functional Theory Calculations of Photoabsorption Spectra in the Vacuum Ultraviolet Region, J. Phys. Chem. A 105 4953–4962 (2001);
Tutorial Validation Checks¶
Check that the main peak of the spectrum has correct position, height and width.
from tutorial_helpers.extract_peak import extract_peak
import numpy as np
run = Run(f"2_Convergence_of_optical_spectra/variable_spacing/run_0.18")
spectrum = run.td.cross_section_vector()
en = spectrum["Energy"].to_numpy()
sf = spectrum["StrengthFunction(1)"].to_numpy()
np.testing.assert_allclose(extract_peak(en, sf, order=1), (13.86, 0.62996, 2.67809), rtol=0.01)
run = Run(f"2_Convergence_of_optical_spectra/variable_spacing/run_0.24")
spectrum = run.td.cross_section_vector()
en = spectrum["Energy"].to_numpy()
sf = spectrum["StrengthFunction(1)"].to_numpy()
np.testing.assert_allclose(extract_peak(en, sf, order=1), (13.86, 0.63098, 2.63694), rtol=0.01)
run = Run(f"2_Convergence_of_optical_spectra/variable_radius/run_3.5")
spectrum = run.td.cross_section_vector()
en = spectrum["Energy"].to_numpy()
sf = spectrum["StrengthFunction(1)"].to_numpy()
np.testing.assert_allclose(extract_peak(en, sf, order=1), (13.86, 0.63135, 2.63576), rtol=0.01)
run = Run(f"2_Convergence_of_optical_spectra/variable_radius/run_7.5")
spectrum = run.td.cross_section_vector()
en = spectrum["Energy"].to_numpy()
sf = spectrum["StrengthFunction(1)"].to_numpy()
np.testing.assert_allclose(extract_peak(en, sf, order=1), (16.24, 0.64528, 1.50324), rtol=0.01)